Effet de matrice
Les méthodes modernes d'analyse chimique utilisent généralement un phénomène physique : si l'intensité du phénomène dépend de la quantité de certains atomes ou molécules, alors en évaluant le phénomène, on peut connaître la quantité d'atomes ou molécules.

Catégories :
Chimie analytique
Les méthodes modernes d'analyse chimique utilisent généralement un phénomène physique : si l'intensité du phénomène dépend de la quantité de certains atomes ou molécules, alors en évaluant le phénomène, on peut connaître la quantité d'atomes ou molécules. Le cas le plus simple est quand l'intensité mesurée du phénomène I est proportionnelle à la concentration en atomes c :
- c = k·I
où k est le cœfficient d'étalonnage.
Cependant, ceci est faux dans le cas général : l'intensité du signal dépend de nombreux autres paramètres, dont les concentrations des autres atomes. Ainsi, un pourcent de fer ne donne pas la même intensité selon qu'il est dans de l'eau ou dans une roche.
L'influence de l'environnement chimique d'un atome est nommé effets de matrice.
Les effets de matrice se rencontrent :
- avec une torche à plasma
- en spectrométrie de fluorescence X
- avec une microsonde de Castaing
- …
Absorption
Un exemple typique de l'effet de matrice est l'absorption des rayonnement. En effet, les méthodes physiques utilisent généralement la mesure d'un rayonnement, soit émis par l'échantillon, soit transmis ou réfléchi par lui. Généralement, les rayonnements sont absorbés suivant une loi de Beer-Lambert :
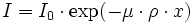
où
- I0 est l'intensité d'origine du signal ;
- I est l'intensité après absorption par l'échantillon ;
- μ est le cœfficient d'absorption massique, qui dépend de la longueur d'onde λ du rayonnement et de la composition chimique de l'échantillon ;
- ρ est la masse volumique de l'échantillon ;
- x est la distance parcourue dans la matière.
Le cœfficient d'absorption μ suit généralement une loi des masses :
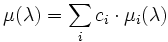
où
- ci est la concentration massique de l'élément i ;
- μi est le cœfficient d'absorption massique de l'élément i (les valeurs sont tabulées) ; les μi ne sont généralement pas continus sur tout le spectre, mais présentent des discontinuités, correspondant aux transitions électroniques des atomes.
Donc, si on suppose que l'intensité I0 ne dépend que de la quantité de l'élément auquel on s'intéresse, l'intensité mesurée I dépend de plus de :
- du chemin x que parcourt le rayonnement dans l'échantillon, et surtout de l'épaisseur de l'échantillon etde l'angle d'incidence du rayonnement ;
- des teneurs en autres éléments (ci) ;
- de la masse volumique ρ de l'échantillon.
Surexcitation
Occasionnellement, les atomes de l'échantillons émettent leurs propres rayonnements (fluorescence) ; ces rayonnements peuvent à leur tour exciter les autres atomes et par conséquent s'ajouter au rayonnement incident. Occasionnellement, ce phénomène n'intervient pas (surtout quand le rayonnement fluorescent n'influe pas sur le phénomène physique de mesure).
Effet chimique
Les liaisons chimiques concernent les électrons périphériques des atomes. Ainsi, les électrons périphériques d'un atome n'ont pas les mêmes niveaux d'énergie selon que l'atome est isolé, fait partie d'une molécule (liaison covalente) ou d'un cristal (liaison ionique, liaison métallique).
Si l'atome est ionisé, cela influe aussi sur les niveaux d'énergie de ces électrons périphériques.
Si les phénomènes physiques mettent en œuvre les électrons périphériques des atomes, ou si l'énergie des rayonnement est de l'ordre de grandeur des énergies de liaison des électrons périphériques, alors le degré d'oxydation ou les liaisons chimiques des atomes peuvent influer sur la mesure, soit sur l'intensité mesurée, soit sur le décalage en énergie des raies mesurées.
En spectrométrie de fluorescence X
La spectrométrie de fluorescence X concerne les électrons de cœur, la méthode n'est par conséquent pas sensible aux liaisons chimiques et au degré d'oxydation (à l'exception notable de l'aluminium et du soufre). On ne considère par conséquent que l'absorption et la fluorescence secondaire (surexcitation).
Ces phénomènes ont été modélisés par J. Sherman en 1955, selon les travaux de Glocker (1930), Castaing (1950) et Gillam et Heal (1952) ; l'équation de Sherman fut corrigée en 1966 part T. Shiraiwa et N. Fujino.
Depuis les années 1980, on dispose d'une puissance de calcul informatique suffisante à un coût raisonnable (micro-ordinateur) servant à corriger les effets de matrice de manière théorique, avec la méthode dites des «paramètres fondamentaux» : cœfficients d'absorption tabulés, rendement de fluorescence… Jusque là, on utilisait des méthodes d'approximation, en utilisant une équation approchée, simplifiée, comme par exemple la formule de Lachance-Traill :
- Ci = mi×Ii× (1 + ∑j≠i αij×Ci)
où
- i est l'élément auquel on s'intéresse, les j sont l'ensemble des autres éléments de l'échantillon ;
- Ci est la concentration de l'élément i ;
- Ii est l'intensité de la raie de l'élément i qu'on mesure ;
- les αij sont les «cœfficients inter-élément» ;
- dans la théorie, ce ne sont pas des constantes, il fluctuent d'un échantillon à l'autre, ils dépendent eux-mêmes de l'ensemble des concentrations ; cependant, pour des variations de concentrations modérées (même type de matériau analysé), ils fluctuent peu ;
- si on dilue l'échantillon, par exemple en faisant une perle fondue, on restreint la gamme de concentrations sur laquelle on travaille, par conséquent on renforce l'hypothèse d'invariabilité des alphas ;
- si on ajoute un élément lourd comme le lanthane dans la perle, on fait tendre les alphas vers 0, mais on rend difficilement mesurables les raies peu intenses (éléments légers ou faibles concentrations) ; cette méthode n'est de fait plus utilisée.
Les alphas sont par conséquent désormais déterminés de manière théorique à partir des paramètres fondamentaux[1]. Ils étaient jusque là reconnus comme fixes et déterminés :
- par régression, ce qui nécessitait un nombre important d'étalons (si on mesure n éléments, il fallait au moins n² étalons avec des variations relatives de concentrations) ;
- de manière théorique, à partir d'un «échantillon moyen» (méthode de Broll-Tertian[2]).
On a aussi utilisé des équations faisant intervenir l'intensité, ou bien des cœfficients de second ordre[3].
Annexes
Bibliographie
- X-Ray Spectroscopy 12, pp 30–37, 1983
- R. Rousseau, J. A. Boivin, The Fundamental Algorithm : A Natural Extension of the Sherman Equation, Part I : Theory, The Rigaku Journal vol. 15 n° 1, pp 13–28, 1998 [3]
- R. Rousseau, The Fundamental Algorithm : An Exhaustive Study of the Claisse-Quentin Algorithm and the Tertian and Lachance Identities, Part II : Application, The Rigaku Journal vol. 15 n° 2, pp 14–26, 1998 [4]
Références
- R. Rousseau, J. A. Boivin, The Fundamental Algorithm : A Natural Extension of the Sherman Equation, Part I : Theory, The Rigaku Journal vol. 15 n° 1, pp 13–28, 1998 [1]
- X-Ray Spectroscopy 12, pp 30–37, 1983
- R. Rousseau, The Fundamental Algorithm : An Exhaustive Study of the Claisse-Quentin Algorithm and the Tertian and Lachance Identities, Part II : Application, The Rigaku Journal vol. 15 n° 2, pp 14–26, 1998 [2]
Recherche sur Google Images : |
"d'aérosol d'échantillon au" L'image ci-contre est extraite du site iramis.cea.fr Il est possible que cette image soit réduite par rapport à l'originale. Elle est peut-être protégée par des droits d'auteur. Voir l'image en taille réelle (854 × 458 - 44 ko - jpg)Refaire la recherche sur Google Images |
Recherche sur Amazone (livres) : |
Voir la liste des contributeurs.
La version présentée ici à été extraite depuis cette source le 30/11/2010.
Ce texte est disponible sous les termes de la licence de documentation libre GNU (GFDL).
La liste des définitions proposées en tête de page est une sélection parmi les résultats obtenus à l'aide de la commande "define:" de Google.
Cette page fait partie du projet Wikibis.
 Accueil
Accueil Recherche
Recherche Début page
Début page Contact
Contact Imprimer
Imprimer Accessibilité
Accessibilité