Spectrométrie de masse / Spectromètre de masse
La spectrométrie de masse est une technique physique d'analyse servant à détecter et d'identifier des molécules d'intérêt par mesure de leur masse, et de caractériser leur structure chimique.

Catégories :
Spectrométrie de masse - Chimie analytique
Définitions :
- méthode qui combine les techniques de chromatographie de gaz liquide avec la spectrométrie de masse pour identifier diverses substances dans l'urine. Comme test spécifique, il identifie précisément la présence d'une substance spécifique dans l'échantillon. (source : fr.fifa)
- technique physique donnant la possibilité de grâce à l'analyse des rayonnements de déterminer la masse d'une molécule. (source : sambamara)
La spectrométrie de masse (mass spectrometry ou MS) est une technique physique d'analyse servant à détecter et d'identifier des molécules d'intérêt par mesure de leur masse, et de caractériser leur structure chimique.
Son principe réside dans la séparation en phase gazeuse de molécules chargées (ions) selon leur rapport masse/charge (m/z). La spectrométrie de masse est utilisée dans quasiment l'ensemble des domaines scientifiques : physique, astrophysique, chimie en phase gazeuse, chimie organique, dosages, biologie, médecine...
Structure d'un spectromètre de masse

Le spectromètre de masse, originellement conçu par le britannique Joseph John Thomson, comporte une source d'ionisation suivie d'un ou plusieurs analyseurs qui séparent les ions produits selon leur rapport m/z, d'un détecteur qui compte les ions et augmente le signal, et enfin d'un dispositif informatique pour traiter le signal. Le résultat obtenu est un spectre de masse représentant les rapports m/z (ou m/q, q représentant la charge) des ions détectés selon l'axe des abscisses et l'abondance relative de ces ions selon l'axe d'ordonnées.
Le spectromètre de masse se compose par conséquent de quatre parties :
- Le dispositif d'introduction de l'échantillon : l'échantillon peut être introduit directement dans la source, sous forme gazeuse, liquide (infusion directe) ou solide (canne d'introduction directe, dépôt sur plaque MALDI, ... ) ou encore par l'association à une méthode séparative (chromatographie en phase liquide, chromatographie en phase gazeuse, électrophorèse capillaire, ... ).
- La source d'ionisation : elle consiste à vaporiser les molécules ainsi qu'à les ioniser. Une source d'ionisation est parfois utilisée soit en mode positif pour étudier les ions positifs, soit en mode négatif pour étudier les ions négatifs. Plusieurs types de sources existent et sont utilisés selon le résultat recherché et des molécules analysées.
- L'ionisation électronique (EI), l'ionisation chimique (CI) et la désorption-ionisation chimique (DCI)
- Le bombardement par atomes rapides (FAB), atomes métastables (MAB) ou ions (SIMS, LSIMS)
- Le couplage plasma inductif (ICP)
- L'ionisation chimique à pression atmosphérique (APCI) et la photoionisation à pression atmosphérique (APPI)
- L'électronébulisation ou électrospray (ESI)
- La désorption-ionisation laser assistée par matrice (MALDI), activée par une surface (SELDI) ou sur silicium (DIOS)
- L'ionisation-désorption par interaction avec espèces métastables (DART)
- L'analyseur : il sépare les ions selon leur rapport masse/charge (m/z). Il existe des analyseurs basse résolution : le quadripôle ou quadrupôle (Q), le piège à ions 3D (IT) ou linéaire (LIT), et des analyseurs haute résolution, servant à mesurer la masse exacte des analytes : le secteur magnétique couplé à un secteur électrique, le temps de vol (TOF), la résonance cyclotronique ionique à transformée de Fourier (FTICR) et l'Orbitrap. Ces analyseurs peuvent être couplés entre eux pour réaliser des expériences de spectrométrie de masse en tandem (MS/MS). Généralement, un premier analyseur sépare les ions, une cellule de collision sert à fragmenter les ions, et un second analyseur sépare les ions fragments. Certains analyseurs, comme les pièges à ions ou le FT-ICR, forment plusieurs analyseurs en un et permettent de fragmenter les ions et d'analyser les fragments directement.
- Le détecteur et dispositif de traitement : le détecteur transforme les ions en signal électrique. Plus les ions sont nombreux, plus le courant est important. Qui plus est , le détecteur augmente le signal obtenu pour qu'il puisse être traité informatiquement.
A quoi sert la spectrométrie de masse ?
- Identification :
- Suivant
le type d'ionisation utilisé, un spectre de masse peut être caractéristique d'une molécule. Ainsi en le comparant avec des banques de spectres, il est envisageable d'identifier la molécule.
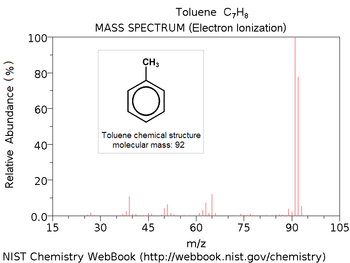 Spectre du toluène en banque de spectres par ionisation électronique
Spectre du toluène en banque de spectres par ionisation électronique
- Lors de l'utilisation d'un analyseur haute résolution (TOF, secteur magnétique, FTICR, Orbitrap), la spectrométrie de masse sert à mesurer avec précision la masse mono-isotopique d'un ion et d'en déduire sa formule brute.
- Suivant
- Analyse structurale :
- La parité de la masse mesurée dépend de la parité du nombre d'atomes d'azote que possède une molécule (règle de l'azote).
- Chaque atome possède un ou plusieurs isotopes qui sont de masses différentes par définition. Ainsi, la proportion de chaque isotope observé sur un spectre de masse, c'est-à-dire le massif isotopique, est caractéristique de la présence de certains atomes et de leur nombre dans l'ion mesuré (en particulier les éléments Cl, et Br, qui présentent des isotopes M et M+2 en quantité notable).
- Les ions peuvent se fragmenter dans un spectromètre de masse : dans la source d'ionisation, dans l'analyseur ou dans une cellule de collision. Comme les fragmentations respectent des lois précises de chimie en phase gazeuse, l'étude de ces fragments sert à déterminer la structure des ions.
- Quantification :
- Un spectromètre de masse est un détecteur universel et particulièrement sensible. Sa gamme linéaire va de 3 à 7 ordres de grandeur, d'où la possibilité d'obtenir une quantification fiable sur un domaine large.
- Imagerie :
- L'analyse point par point d'une surface par spectrométrie de masse avec ionisation correcte (MALDI, SIMS, DESI) sert à générer des images ioniques, représentant la répartition de chaque ion issu de cette surface. Cette technique d'imagerie est particulièrement utilisée pour la recherche de biomarqueurs (identification dans une coupe de tissu de composés spécifiques d'une région définie).

La source d'ionisation
Les ionisations EI et CI, qui nécessitent un certain niveau de vide, sont préférentiellement utilisées en couplage avec la chromatographie en phase gazeuse (la CI fonctionnant à partir d'une source EI). Par contre, les deux sources à pression atmosphérique (électrospray et APCI) dites à "ionisation douce", sont essentiellement utilisées en couplage avec la chromatographie en phase liquide.
L'ionisation électronique (EI)

Des électrons émis par un filament rencontrent les molécules qui entrent dans la source : lors de la rencontre, si l'énergie cinétique des électrons est suffisante, un électron est arraché de la molécule M, la transformant en un ion radical M+o. Ce dernier peut ensuite se fragmenter suivant son énergie interne. L'EI conduit ainsi à un spectre assez apporté, avec de nombreux fragments, particulièrement riche en informations structurales.
L'ionisation chimique (CI)

En plus du système EI ci-dessus, un gaz réactif est introduit dans la source et ionisé par impact électronique. S'ensuit une série de réactions qui donne naissance à des ions pouvant réagir avec les molécules d'analyte arrivant dans la source. Ce type de réactions ions-molécules produit essentiellement (en mode positif) des ions [MH] +, et [M+adduit+H]+, donnant la possibilité d'ainsi d'accéder à la masse moléculaire de l'analyte.
Le méthane, l'isobutane et l'ammoniac sont parmi les gaz d'ionisation chimique les plus utilisés.
Pour la détection de molécules globalement électronégatives, comportant des parties halogénées ou fluorogénées, on peut faire appel à l'ionisation chimique négative. Le principe est de charger négativement ces molecules en les bombardant d'électrons qui seront capturés par les atomes électroattracteurs. Du fait de la forte probabilité de capture de l'electron, ce type d'ionisation peut être 1000 fois plus sensible que l'ionisation chimique positive[1]
L'ionisation par bombardement d'atomes rapides (FAB)
Elle permet d'analyser des molécules non vaporisables sous vide (grosses molécules biologiques). L'ionisation est effectuée par expulsion en phase vapeur des ions contenus dans un échantillon liquide suite à un bombardement d'atomes rapides (Ar ou Xe). Les molécules ainsi ionisées n'ont pas énormément d'énergie interne, la fragmentation est par conséquent faible mais l'ion moléculaire est aisément reconnaissable et la masse moléculaire est facile à déterminer. L'échantillon est mélangé en solution à une matrice liquide non volatile (glycérol, thioglycérine, alcool m-nitrobenzylique). Un faisceau à haute énergie (de l'ordre de 4 à 10 keV) d'atomes neutres (Ar ou Xe) est envoyé sur l'échantillon et la matrice dans la chambre de collision causant ainsi les phénomènes de désorption et d'ionisation. Les ions préexistants en solution sont expulsés en phase gazeuse et accélérés vers l'analyseur.
L'ionisation par électronébulisation (électrospray) (ESI)

Son principe est le suivant : à pression atmosphérique, les gouttelettes de solutés sont constituées à l'extrémité d'un fin capillaire porté à un potentiel élevé. Le champ électrique intense leur confère une densité de charge importante. Sous l'effet de ce champ et grâce à l'assistance éventuelle d'un courant d'air co-axial, l'effluent liquide est transformé en nuage de fines gouttelettes (spray) chargées suivant le mode d'ionisation. Sous l'effet d'un second courant d'air chauffé, les gouttelettes s'évaporent progressivement. Leur densité de charge devenant trop importante, les gouttelettes explosent en libérant des microgouttelettes constituées de molécules protonées ou déprotonées de l'analyte, porteuses d'un nombre de charges variable.
Les ions ainsi constitués sont ensuite guidés avec potentiels électriques appliqués sur deux cônes d'échantillonnage successifs jouant le rôle de barrières avec les parties en aval maintenues sous un vide poussé (<10-5 Torr). Durant ce parcours à pression élevée, les ions subissent de multiples collisions avec les molécules de gaz et de solvant, ce qui complète leur désolvatation. En faisant fluctuer les potentiels électriques appliqués dans la source il est envisageable de provoquer des fragmentations plus ou moindres.
L'avantage de cette méthode d'ionisation comme pour l'APCI est l'obtention d'ions multichargés, pour les macromolécules, polymères. Elle permet d'autre part de générer une ionisation "douce" : des ions moléculaires sont constitués en majorité.
L'ionisation chimique à pression atmosphérique (APCI)
Les échantillons liquides sont directement introduits dans un nébuliseur pneumatique. Sous l'effet d'un jet d'air ou d'azote, le liquide est transformé en fin brouillard. Un chauffage assure la désolvatation des composés. Ces derniers sont ensuite ionisés chimiquement à pression atmosphérique : généralement, la phase mobile vaporisée joue le rôle de gaz d'ionisation et les électrons sont obtenus à partir de décharges d'électrode couronne. L'ionisation des composés est particulièrement favorisée lors de ces techniques car la fréquence des collisions est élevée à pression atmosphérique.
L'APCI est une technique analogue à l'ionisation chimique (CI), elle fait appel à des réactions ions-molécules en phase gazeuse, mais à pression atmosphérique et conduit principalement à la formation d'ions [MH]+ ou [M-H]-.
La désorption-ionisation laser assistée par matrice (MALDI)

Un faisceau laser pulsé est utilisé, le plus souvent dans le domaine des ultraviolets, pour désorber et ioniser un mélange matrice/échantillon cocristallisé sur une surface métallique, la cible.
Les molécules de matrice absorbent l'énergie transmise par le laser sous forme de photons UV, s'excitent et s'ionisent. L'énergie absorbée par la matrice provoque sa dissociation et son passage en phase gazeuse. Les molécules de matrice ionisées transfèrent leur charge à l'échantillon. L'expansion de la matrice entraîne l'échantillon au sein de la phase gazeuse dense où il va finir de s'ioniser.
L'ionisation de l'échantillon a par conséquent lieu soit dans la phase solide avant la désorption, soit par transfert de charge lors de collisions avec la matrice excitée après désorption. Elle conduit à la formation d'ions monochargés et multichargés de type [M+nH]n+, avec une nette prépondérance pour les monochargés.
L'analyseur
Les analyseurs se différencient par leur principe de mesure du rapport m/z des ions, qui est :
- la dispersion des ions, fondée sur leur moment ou leur énergie cinétique (instruments à secteur magnétique ou électrique)
- la séparation dans le temps, fondée sur la vitesse des ions (TOF)
- la transmission des ions traversant un champ électrodynamique (quadripôle)
- le mouvement périodique dans un champ magnétique ou électrodynamique (pièges ou trappes à ions)
L'analyseur quadripolaire
Un quadripôle (ou quadrupôle) est constitué de quatre électrodes parallèles de section hyperbolique ou cylindrique. Les électrodes opposées distantes de 2r0 sont reliées entre elles et soumises au même potentiel.

Les électrodes adjacentes sont portées à des potentiels de même valeur, mais opposés de sorte que l'écart de potentiel soit égal à φ0.
Ce potentiel φ0 résulte de la combinaison de tensions, l'une continue (U) l'autre alternative (V) de haute fréquence f : 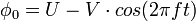
En appliquant cette différence de potentiel entre chaque paire d'électrodes, il se crée un champ électrique quadripolaire. Un point de coordonnées (x, y, z) localisé dans le champ électrique sera alors soumis au potentiel : 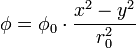

La trajectoire d'un ion pénétrant dans le quadripôle sera par conséquent uniforme selon l'axe z et décrite par les équations de Mathieu selon les deux autres axes. Il est envisageable de définir suivant les valeurs U et V des zones de stabilité telles que les coordonnées x et y de l'ion restent strictement inférieures à r0. L'une d'entre elles est exploitée en spectrométrie de masse (voir figure) (Les ions qui se trouvent dans cette zone auront par conséquent une trajectoire stable dans le quadripole et seront détectés). En gardant constant le rapport U/V, on obtient une droite de fonctionnement de l'analyseur. Un balayage de U avec U/V constant permet l'observation successive de l'ensemble des ions dont la zone de stabilité est coupée par la droite de fonctionnement. La résolution entre ces ions est d'autant plus grande que la pente de la droite est élevée.

En l'absence de tension continue, l'ensemble des ions de rapports m/z supérieurs à celui fixé par la valeur de V appliquée auront une trajectoire stable (x et y < r0), le quadripôle est alors dit transparent et sert de focalisateur d'ions.
Les principaux avantages du spectromètre quadripolaire résident dans sa souplesse d'utilisation, sa résolution unitaire sur toute sa gamme de masse, sa vitesse de balayage satisfaisante, mais aussi son adaptabilité à différentes interfaces donnant la possibilité de le couplage avec la chromatographie gazeuse ou liquide.
L'analyseur octopolaire

Semblable à l'analyseur quadripolaire mais avec 8 électrodes, cet analyseur sert seulement à la focalisation des ions.
Le piège ionique quadripolaire ("trappe d'ions")

C'est un piège ionique où la préparation, l'analyse et la détection des ions s'effectuent dans un même espace, suivant des séquences temporelles successives.
Le piège est constitué de trois électrodes à section hyperbolique : une électrode annulaire encadrée par deux électrodes-chapeaux (d'entrée et de sortie) qui forment les calottes supérieure et inférieure du système. Une tension en radiofréquence 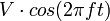 combinée ou non à une tension continue U est appliquée entre l'électrode centrale et les deux électrodes calottes. [2] Le champ résultant est alors tridimensionnel.
combinée ou non à une tension continue U est appliquée entre l'électrode centrale et les deux électrodes calottes. [2] Le champ résultant est alors tridimensionnel.
Les domaines de stabilité des ions sont à nouveau déterminés par les équations de Mathieu. Celui exploité est défini tel que quand les ions en sortent, leur trajectoire radiale reste stable au contraire de celle selon l'axe des z. Un balayage de l'amplitude de la radiofréquence V entraînera par conséquent l'expulsion des ions piégés selon cet axe, vers le détecteur. [3] [4] Les trajectoires stables des ions, au sein du champ quadripolaire résultant sont tridimensionnelles, en forme de huit.
Le temps de vol
L'analyseur à temps de vol consiste à mesurer le temps que met un ion, accéléré préalablement par une tension, à parcourir une distance donnée. Le rapport masse sur charge est directement mesurable à partir du temps de vol.

Un analyseur à temps de vol se compose d'une zone d'accélération où est appliquée la tension accélératrice, et d'une zone nommée tube de vol, libre de champ. Les ions accélérés pénètrent dans le tube de vol libre de tout champ. La séparation des ions ne va par conséquent dépendre que de la vitesse acquise lors de la phase d'accélération. Les ions de rapport m/z le plus petit parviendront au détecteur les premiers. Pour chaque groupe d'ions de même rapport m/z, un signal est enregistré au niveau du détecteur sous la forme d'une fonction temps/intensité.
Ce mode de détection comporte cependant certaines limitations en termes de résolution : ainsi deux ions semblables, de même vitesse d'origine, mais situés à deux points différents, entreront dans le tube de vol à des vitesses et des temps différents. Celui le plus loin du détecteur à l'origine sera accéléré plus longtemps et aura par conséquent un temps de vol plus court, d'où une dispersion en temps et en énergie. Le mode réflectron sert à pallier ce phénomène.

En mode réflectron, un miroir électrostatique impose un champ électrique de direction opposée à celle du champ accélérateur d'origine, et par conséquent du mouvement des ions. Ces derniers voient ainsi leur trajectoire modifiée : ils pénètrent dans le réflectron et en ressortent avec une vitesse longitudinale de sens opposé à leur vitesse d'origine. Les ions les plus énergétiques arrivent les premiers au niveau du réflectron et vont y pénétrer plus profondément, ils seront par conséquent réfléchis dans un temps plus long. De cette façon, l'ensemble des ions de même rapport m/z se trouvent focalisés sur un même plan, le détecteur du réflectron étant positionné sur le plan de focalisation de ces ions. En outre, le réflectron permet d'allonger la distance de vol sans pour tout autant augmenter la taille de l'analyseur : les ions mettent plus de temps pour atteindre le détecteur, et diminuent aussi leur dispersion en temps, la résolution s'en trouve par conséquent largement perfectionnée.
Le FT-ICR

L'analyseur à résonance cyclotronique d'ion se compose d'une cellule ICR (de configuration cubique par exemple) qui comporte surtout six plaques sous tension, isolées les unes des autres.
L'application d'un champ magnétostatique B suivant l'axe z soumet les ions à la force de Lorentz F = ez v ˆ B. Leur mouvement dans le plan (xy) est alors «cyclotronique», c'est-à-dire circulaire uniforme de fréquence f= eB/ (2π. m/z). Les ions sont d'autre part confinés suivant l'axe z par un champ électrostatique imposé par les deux plaques parallèles au plan (Oxy), résultant de l'application d'une tension faible.
Une fois piégés dans la cellule, les ions ont par conséquent la même trajectoire mais pas la même position à un instant déterminé (a).

Il convient par conséquent de donner aux ions de même m/z un mouvement d'ensemble en les mettant en phase, par résonance cyclotronique. Pour cela, les ions m/z sont excités par un champ alternatif de fréquence correspondant à leur fréquence cyclotron : pour exciter l'ensemble des ions d'une certaine gamme de m/z, une tension contenant l'ensemble des fréquences cyclotron correspondantes est imposée. Les ions sont alors accélérés, mis en phase et voient le rayon de leur orbite augmenter (b).
Le courant induit par le mouvement cohérent des ions de même m/z sera mesuré sur les plaques de détection (c) : ce sera une sinusoïde amorti de fréquence cyclotronique. Le courant induit total mesuré sera par conséquent la somme de sinusoïdes amorties des fréquences cyclotroniques correspondant aux ions de m/z excités par résonance. La fréquence cyclotron étant proportionnelle à 1/ (m/z), l'inverse de la transformée de Fourier du courant obtenu permet d'aboutir au spectre de masse en m/z.
Cet analyseur a l'une des meilleures résolutions qui soient (Rs>100 000), par conséquent le spectre MS a une plus grande capacité de pics, ce qui maximise la quantité d'informations pour l'analyse de mélanges complexes. Cependant, la largeur des pics étant proportionnelle à (m/z) ², la résolution est meilleure aux m/z inférieures à 5000 Th. L'excellente précision du FT-ICR sur la mesure de masse (5-10 ppm) lève ou diminue les ambiguïtés sur l'identification des composés. La gamme de masse dépend de la valeur du champ magnétique, elle couvre jusque 27000 Da pour un champ de 7 T. Par contre, la gamme dynamique est assez restreinte, avec 2-3 décades, car cet analyseur par confinement souffre du même défaut que le piège quadripolaire, la cœxistence envisageable d'un nombre limité d'ions. Par conséquent, les pics particulièrement minoritaires dans le spectre de masse présenteront une mesure de masse moins précise. Le FT-ICR permet l'analyse en MS/MS dans la cellule même, avec possibilités variées d'activation des ions et par conséquent de fragmentations sélectives.
L'orbitrap

L'orbitrap se compose d'une électrode creuse, au sein de laquelle est positionnée coaxialement une électrode en forme de fuseau. La forme spécifique de ces deux électrodes permet l'imposition d'un champ électrostatique quadro-logarithmique avec la tension : 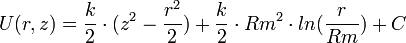 .
.
avec Rm rayon caractéristique de l'électrode centrale, k courbure du champ, et C une constante.
Le champ est surtout quadripolaire suivant l'axe z des électrodes. Les ions sont injectés tangentiellement à l'électrode centrale et piégés autour d'elle par la force électrostatique qui compense les forces centrifuges. Le mouvement des ions se décompose alors ainsi : un mouvement circulaire autour de l'électrode centrale dans le plan (xy) et un mouvement oscillatoire de va-et-vient selon l'axe z. [5] Surtout, les ions d'un m/z donné seront sur la même trajectoire circulaire qui oscille axialement avec une fréquence f. f est indépendante de la vitesse ou de l'énergie des ions et s'exprime comme 1/2π√ (km/z). De la même façon que pour le FT-ICR, le courant induit par ces oscillations permet par une transformée de Fourier d'accéder aux m/z.
La précision des mesures de m/z est spécifiquement bonne (1-2 ppm) et la résolution (jusque 100 000) rivalise avec celle du FT-ICR, d'autant qu'étant proportionnelle à 1/√ (m/z), elle diminue moins vite avec le rapport m/z que dans le cas du FT-ICR. La gamme dynamique est satisfaisante (> 3 décades). [6] L'orbitrap est essentiellement utilisée en spectrométrie de masse en tandem, associée à un piège linéaire.
L'analyseur à secteur magnétique

L'ion est éjecté dans un milieu dans lequel règne un champ magnétique uniforme perpendiculaire au plan de la trajectoire. Du fait de la force de Lorentz, la trajectoire se courbe, et le point d'impact de l'ion (donc sa déviation) sert à connaître sa masse à partir de la charge.
En effet, soit 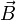 le champ magnétique (dirigeant
le champ magnétique (dirigeant 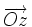 ) de coordonnées
) de coordonnées 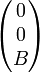 et
et 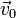 la vitesse d'origine orthogonale à , elle dirige
la vitesse d'origine orthogonale à , elle dirige 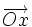 .
.
On a alors : 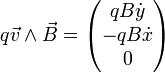 .
.
D'où, en écrivant la relation principale de la dynamique : 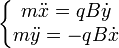 .
.
Soit : 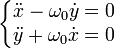 où
où 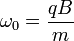 .
.
Posons 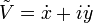 .
.
On a alors 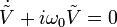 .
.
En résolvant, 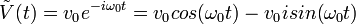 .
.
Et donc : 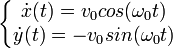
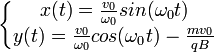 (avec conditions initiales).
(avec conditions initiales).
Il s'agit bien de l'équation paramétrique d'un cercle de rayon 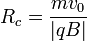 .
.
Le spectromètre mesure ensuite les distances d'impact quand la particule a effectué un demi-cercle. La distance au point d'origine correspond au diamètre par conséquent au double du rayon donné par la dernière formule. La charge de la particule permet par conséquent d'en déduire sa masse.
Le détecteur
Comme les analyseurs et les sources, il existe différents types de détecteurs. Ils sont tous basés sur des principes physiques différents, mais leur rôle reste le même, compter les ions. C'est une partie positionnée sous vide (10-5 - 10-7 Torr).
- Les plaques photographiques sont le détecteur historique. La plaque est enduite d'une émulsion de bromure d'argent, son noircissement donne une valeur relative de l'intensité du flux (quantité d'ions). Cette technique est particulièrement peu sensible.
- Le cylindre de Faraday a pour principe le suivant : le transfert de charge de l'ion est détecté sur une surface conductrice, puis le signal est augmenté. Cette technique est précise mais peu sensible, avec une certaine lenteur de mesure et un bruit de fond important.
- Le multiplicateur d'électrons est le détecteur le plus courant. Le signal est augmenté par la formation d'électrons secondaires avec tubes en verre dopés au plomb (dynode). Il possède une bonne sensibilité, avec une augmentcation forte mais il est moins précis que le cylindre de Faraday. Il a en outre une durée de vie limitée. La galette de microcanaux, autre détecteur, peut être reconnue comme assemblage de multiplicateurs d'électrons.
- Le multiplicateur de photons est dérivé du multiplicateur d'électrons : le signal est augmenté par la formation d'électrons secondaires avec tubes en verre dopés au plomb (dynode). Ceux-ci sont accélérés vers un écran phosphorescent où ils sont convertis en photons. Ces photons sont ensuite détectés par le photomultiplicateur. Il présente une bonne sensibilité, avec augmentcation forte mais le balayage est moins rapide qu'avec un multiplicateur d'électrons.
La spectrométrie de masse en tandem (MS/MS)
Voir aussi : Séquençage par spectrométrie de masse

La spectrométrie de masse en tandem consiste à sélectionner un ion par une première spectrométrie de masse, à le fragmenter, puis à effectuer une deuxième spectrométrie de masse sur les fragments ainsi générés.
Elle peut être réalisée avec nombreux appareils combinant des secteurs magnétiques, électriques, quadripolaires ou des temps de vol, ainsi qu'au sein d'un même analyseur dans le cas d'une trappe d'ions.
Le triple quadripôle

Un triple quadripôle résulte de l'association de deux analyseurs quadripolaires en série, scindés par une cellule de collision fréquemment constituée d'un quadripôle plus court. Cette combinaison de quadripôles sert à travailler en MS simple ou en tandem. Pour réaliser une acquisition en MS, il suffit de n'appliquer qu'une tension alternative à l'un des analyseurs pour le rendre "transparent" comme la cellule de collision, celle-ci ne contenant alors pas de gaz.
Lors d'une acquisition en MS/MS, la cellule de collision est remplie d'un gaz inerte (argon par exemple) sous une pression assez élevée (10 − 2 torr). L'énergie cinétique de l'ion choisi est convertie lors de ses collisions successives en énergie interne. La dissociation de l'ion se réalisera quand son énergie interne sera devenue supérieure à l'énergie d'activation indispensable à la fragmentation. Cette technique de dissociation activée par collision (CAD) peut être augmentée en augmentant l'énergie cinétique des ions choisies par application d'une différence de potentiel entre la source et la cellule de collision.
L'analyse MS/MS peut être menée selon quatre modes différents selon l'information recherchée : le mode descendant est le plus utilisé pour obtenir des informations structurales, les deux modes (ascendant et perte de neutre) sont d'un usage plus restreint et permettent de mettre en évidence des ions ayant des particularités communes. Le quatrième mode (Multiple Reaction Monitoring ou MRM), dérivé du mode descendant, est voué à la quantification.
- en mode descendant, l'ion à étudier est choisi en focalisant le premier analyseur sur son rapport m/z. Les fragments constitués dans la cellule de collision sont scindés par le deuxième analyseur et analysés. Le spectre obtenu présente à la fois l'ion précurseur (ou ion parent) et ses ions fragments (ou ions produits).
- en mode ascendant, le premier analyseur balaie une gamme de masse alors que le deuxième est focalisé sur un seul rapport m/z. L'ensemble des ions générés en source et capables de donner un fragment de même rapport m/z seront par conséquent ainsi détectés.
- en mode perte de neutre, les deux analyseurs balaient une gamme de masse simultanément et avec un décalage de masse constant. Le spectre établi présentera alors l'ensemble des ions parents capables de se fragmenter en générant un neutre de masse égale au décalage imposé.
- en mode MRM, l'ion parent à étudier est choisi par le premier analyseur et fragmenté dans la cellule de collision, comme en mode descendant. Par contre, le second analyseur est focalisé sur l'ion produit. Ce mode de fonctionnement présente une double sélectivité, au niveau des sélections de l'ion parent et de l'ion produit. En outre les deux analyseurs étant fixées à des tensions constantes, la sensibilité de détection est perfectionnée comparé à d'autres modes de balayage, faisant de la MRM un mode de choix pour la quantification.
Piège à ions quadripolaire ("trappe d'ions")
Au sein d'un piège à ions quadripolaire (quelquefois nommé "trappe d'ions" en raison d'une mauvaise traduction de l'anglais "ion trap"), l'analyse en tandem se réalise tout d'abord par sélection d'ions dont la valeur m/z est choisie. Ces ions piégés vont ensuite se fragmenter par collisions (acquisition d'énergie interne, excitation vibrationnelle) avec une tension RF (radiofréquence) correspondant à leur fréquence de résonance, et les ions produits constitués sont à leur tour piégés. Une éjection sélective en masse des ions produits (fragments) peut alors être réalisée en vue de leur analyse. Le gaz de collision est le plus souvent de l'hélium présent en permanence dans le piège et a pour rôle aussi de focaliser les ions aux centre de l'analyseur.
L'obtention d'ions de générations supérieures est envisageable par simple renouvellement du processus (sélection d'un ion produit, fragmentation, sélection d'un ion produit de 2e génération, fragmentation, etc... ). Cette séquence est nommée MSn, n étant le nombre de générations d'ions. Ainsi la MS2 est la MS-MS et ainsi de suite...
Hybride
Associer plusieurs types d'analyseur dans un spectromètre de masse en tandem sert à combiner les points forts des deux types d'analyseurs.
- Quadripôle/temps de vol :
Ces appareils nommés Q-TOF sont constitués d'un double quadripôle (1er analyseur + cellule de collision) et d'un analyseur à temps de vol comme second analyseur. Le quadripôle procure ainsi une grande efficacité au processus MS/MS, alors que le TOF apporte son excellente sensibilité, sa grande rapidité d'analyse et ses résolution et précision en masse bien meilleure sur les ions produits, comparé à une configuration triple quadripôle. Cependant ces instruments sont limités par la faible gamme dynamique du TOF.
- Quadripôle/piège à ions quadripôlaire :
Cette combinaison permet d'éviter les problèmes de charge d'espace associés aux pièges à ions. Le piège apporte une meilleure sensibilité et une vitesse d'analyse plus rapide. En outre, comparé à un piège simple, cette combinaison autorise l'ensemble des modes d'acquisition MS/MS du triple quadripôle (perte de neutre et mode ascendant).
- Piège à ions quadripôlaire/temps de vol :
L'association d'un piège à ions et d'un TOF permet d'accéder à une analyse structurale particulièrement poussée par MSn grâce au piège, mais présente aussi une grande précision en masse sur les ions précurseurs et produits pour la détermination des formules brutes, complétant ainsi l'identification. Cet appareil hybride est ainsi essentiellement dédié à l'identification ainsi qu'à l'analyse structurale.
- Piège à ions/FT-ICR ou Piège à ions/Orbitrap :
L'objectif de ce genre de couplage est d'obtenir une précision en masse et une résolution, qui soient toujours meilleures qu'avec un TOF comme deuxième analyseur, et permettent l'établissement de formules brutes sans ambigüité et par conséquent une identification facilitée. Ces appareils représentent cependant un investissement bien supérieur, comparés à un piège à ions/temps de vol.
Voir aussi
Liens externes
- La spectrométrie de masse pour l'analyse chimique et biologique : Un article Ecole Normale Supérieure - DGESCO (plusieurs liens sur le sujet)
- Cours d'Olivier Laprévote en spectrométrie de masse
- Aperçu des sites pertinents sur la Spectrométrie de masse (anglais)
- Site de l'académie de Nancy et Metz proposant des cours sur la chimie organique dont la spectroscopie de masse.
Références
- Gas Chromatography and mass spectrometry : a practical guide, Kitson, Larsen & McEwen, academic press, 1996
- Lawson, G. ; Todd, J. F. J. ; Bonner R. F., Dyn. Mass Spectrom. 1975, 4, 39
- Dawson, P. H. ; Whetten, N. R., J. Vac. Sci. Technol. 1968, 5, 11
- Campana, J. E., Int. J. Mass Spectrom. Ion Phys. 1980, 33, 101
- Hu, Q. ;Noll, R. J. ; Li, H. ;Makarov, A. ; Hardman, M. ; Cooks, G., J. Mass Spectrom. 2005 40 (4), 430
- Makarov, A. ; Denisov, E. ; Kholomeev, A. ; Balschun, W. ; Lange, O. ; Strupat, K. ; Horning, S. Anal Chem. 2006 78 (7), 2113
Recherche sur Google Images : |

"Spectrométrie de masse" L'image ci-contre est extraite du site dunod.com Il est possible que cette image soit réduite par rapport à l'originale. Elle est peut-être protégée par des droits d'auteur. Voir l'image en taille réelle (250 × 352 - 14 ko - jpg)Refaire la recherche sur Google Images |
Recherche sur Amazone (livres) : |
Voir la liste des contributeurs.
La version présentée ici à été extraite depuis cette source le 30/11/2010.
Ce texte est disponible sous les termes de la licence de documentation libre GNU (GFDL).
La liste des définitions proposées en tête de page est une sélection parmi les résultats obtenus à l'aide de la commande "define:" de Google.
Cette page fait partie du projet Wikibis.
 Accueil
Accueil Recherche
Recherche Début page
Début page Contact
Contact Imprimer
Imprimer Accessibilité
Accessibilité